John Manak
Research summary
Genomics, genetics, neurobiology
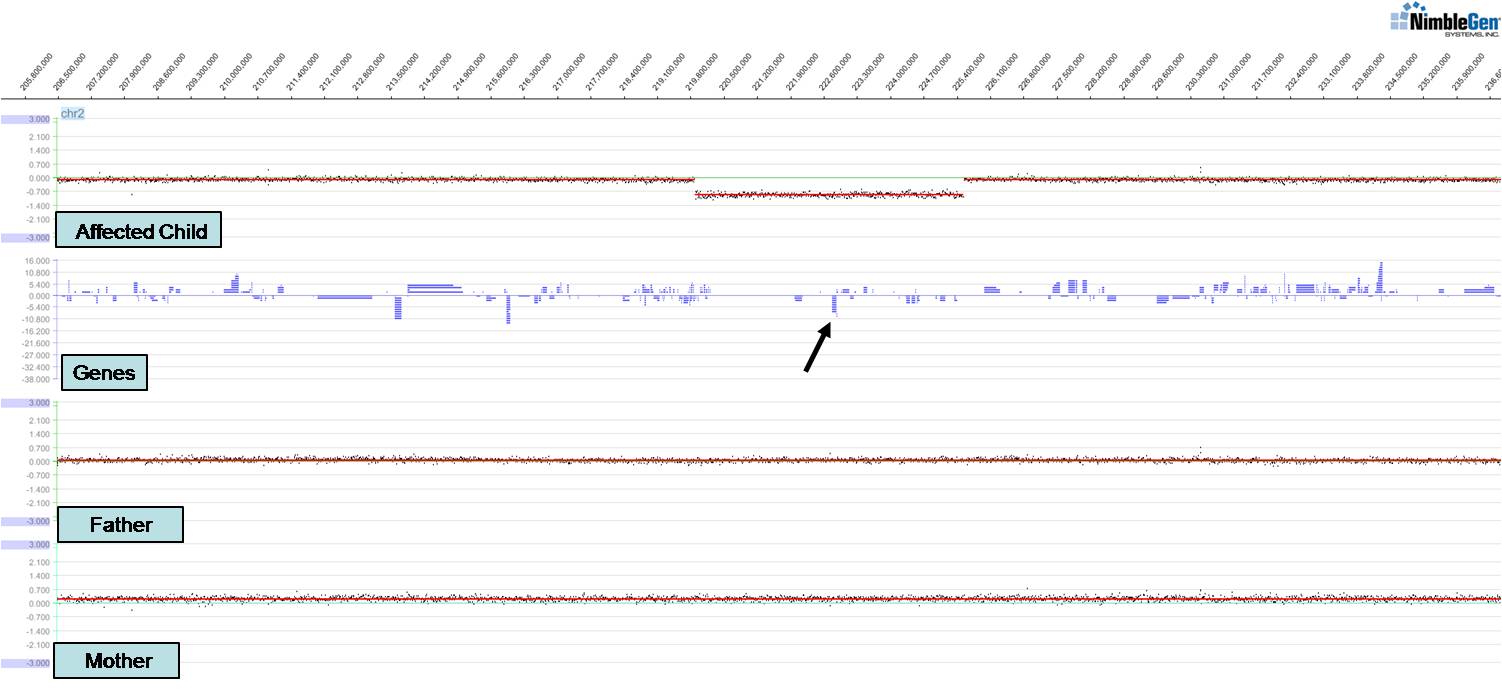
Work in my laboratory focuses on understanding human genetic disorders by first identifying gene mutations associated with those disorders and then modeling the disorder in relevant organisms. We have identified novel genes and chromosomal structural variants (see figure) associated with a variety of congenital anomalies including spina bifida, branchio-oto-renal syndrome, renal agenesis, and cleft lip and/or palate. These studies have led to publications in Hum Mol Genet, Hum Genet, Genetics (invited and highlighted articles), and Am J Hum Genet amongst others. Of particular interest is a new clefting and craniofacial patterning gene we identified, ISM1. We have found that knockdown of ISM1 in frogs inhibits migration of cranial neural crest cells, the key cells that make up the structure of the face, thus leading to severe craniofacial anomalies including clefts. We have also studied the fly homolog of the C-MYB proto-oncogene (Dm-Myb) where we discovered a new function for Myb in separating and stabilizing chromatin neighborhoods associated with topologically associating domains (TADs); loss of Dm-Myb results in loss of H3K27me3 repressive domains, leading to robust derepression of large numbers of genes in the genome. Interestingly, Myb is the primary DNA-binding factor that binds to TAD chromatin loop anchors in flies and may also be one of the key anchor proteins working alongside CTCF in humans. These studies were published in Nature, Nature Cell Biol, PNAS, and Cell Reports amongst others.
A major focus of the laboratory is understanding the genetic basis of epilepsy, using a fruit fly model of an epilepsy-ataxia syndrome. We have shown that fly PRICKLE mutants exhibit a remarkably similar syndrome to humans carrying PRICKLE mutations (early-onset, progressive, myoclonic/tonic-clonic seizures as well as ataxia) and have identified enhanced axonal transport defects as one cause of the seizure phenotype. Recently, we have shown that activation of the glial innate immune response (IIR) leads to increased neuronal cell death which is responsible for exacerbating the seizure phenotype and that increases in neuronal oxidative stress ultimately lead to activation of the IIR. We thus provide the first direct genetic evidence that oxidative stress and the glial IIR can lead to the progression of epilepsy. These studies were published in PNAS, Am J Hum Genet, PLoS Genetics, Ann Clin Transl Neurol, and Cell Reports amongst others. Current studies are aimed at determining whether we can repurpose well-tolerated antioxidant and anti-inflammatory drugs as a new class of anti-epileptic drugs (AEDs). We are also exploring how the glia are activated in our seizure mutant; for example, are damage-associated molecular patterns (DAMPs) the culprit, and which ones? Additionally, we are exploring how the glial IIR is able to promote neuronal cell death, thus exacerbating the seizure phenotype.
Selected image
- Genetics
- Neurobiology
